Ayer tuve la suerte de poder participar y ayudar en la definició de un treball de recerca de bachillerato que puede resultar muy, muy interesante. Además, entronca con una de las ideas que veo con más recorrido: utilizar las herramientas e información disponible en las bases de datos públicas para hacer ciencia en los institutos (y, por tanto, enseñar y aprender conceptos y, lo que es igual o más importante, la manera de funcionar de la ciencia).
El treball de recerca utilizará los datos y las herramientas que pone a disposición de todo el mundo el NCBI. En este caso en concreto parece que se centrarán en la comparación masiva de secuencias de nucleótidos o de aminoácidos para responder a diferentes preguntas. Me reservo la sorpresa para más adelante. Tomároslo como un teaser sin spoilers.
Aprovecharé la ocasión para dejaros con un protocolo de cómo realizar comparaciones masivas de las secuencias que creáis más convenientes.
El treball de recerca utilizará los datos y las herramientas que pone a disposición de todo el mundo el NCBI. En este caso en concreto parece que se centrarán en la comparación masiva de secuencias de nucleótidos o de aminoácidos para responder a diferentes preguntas. Me reservo la sorpresa para más adelante. Tomároslo como un teaser sin spoilers.
Aprovecharé la ocasión para dejaros con un protocolo de cómo realizar comparaciones masivas de las secuencias que creáis más convenientes.
Buscar una secuencia
En el NCBI, entre otras muchísimas cosas, podemos buscar tanto secuencias de nucleótidos como proteínas. Utilizaré como ejemplo mi vieja amiga MKP-1 (o DUSP-1). Durante cuatro años fuimos íntimos; supongo que ella ya no se acordará de mí (snif). La frase perenne.
Para buscar su secuencia de nucleótidos, utilizaremos la base de datos Nucleotide. Ponemos «MKP-1» en la barra de búsqueda. Intro. Et voilá:
Para buscar su secuencia de nucleótidos, utilizaremos la base de datos Nucleotide. Ponemos «MKP-1» en la barra de búsqueda. Intro. Et voilá:
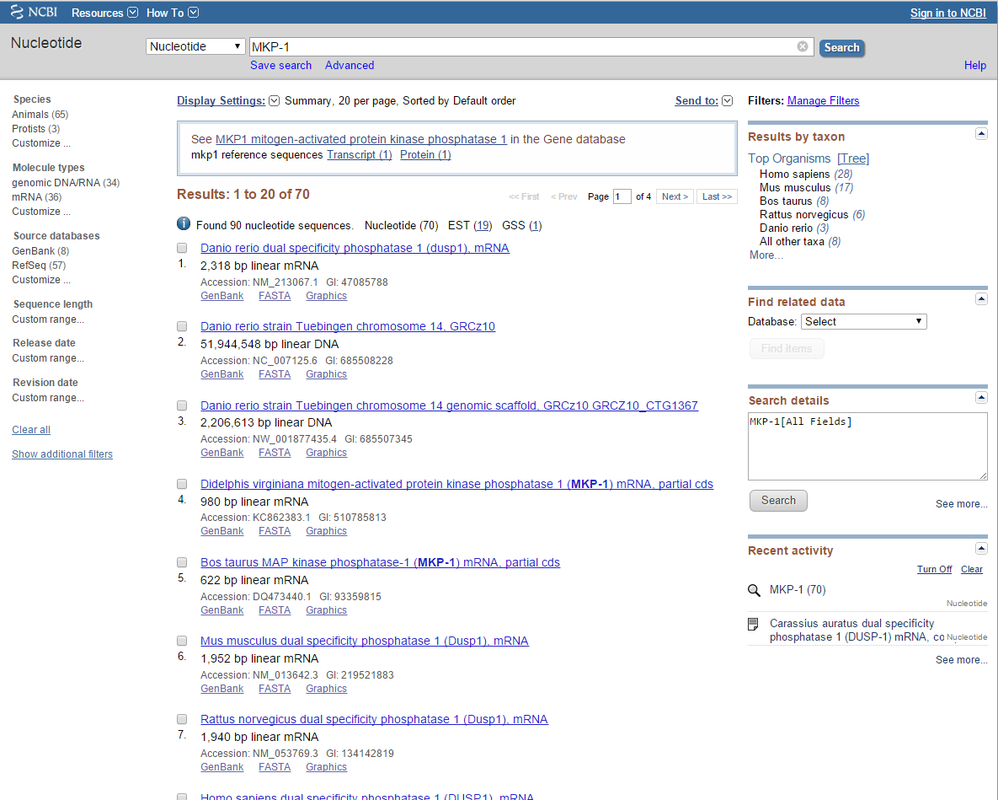
Nos devuelve 70 secuencias que encajan con nuestra búsqueda.Debemos fijarnos bien en qué nos encontramos en cada resultado. En este ejemplo podemos encontrar tanto mRNAs de 2.000 pares de bases (resultado 1) como cromosomas de 51 millones de pares de bases (resultado 2).
Si la cantidad de resultados es abrumadora (y suele serlo), podemos acotar, a la izquierda, por especie, por tipo de molécula (no es lo mismo utilizar DNA que RNA); a la derecha podemos filtrar por taxón (es decir, de nuevo por especie).
Podemos acceder a la secuencia que nos interese clicando sobre su nombre. Accederemos así a toda la información sobre la secuencia. Lo cual siempre es interesante. Sin embargo, si queremos realizar una búsqueda rápida, lo mejor es clicar sobre la opción «FASTA» que encontramos debajo de los Accession numbers i gi (éstos también nos servirían para hacer la comparación).
Inciso: La búsqueda de secuencias proteicas (de aminoácidos) funciona de la misma manera, aunque, evidentemente, la búsqueda la tenemos que realizar en la base de datos Protein.
Volvamos a nuestro FASTA.
Si la cantidad de resultados es abrumadora (y suele serlo), podemos acotar, a la izquierda, por especie, por tipo de molécula (no es lo mismo utilizar DNA que RNA); a la derecha podemos filtrar por taxón (es decir, de nuevo por especie).
Podemos acceder a la secuencia que nos interese clicando sobre su nombre. Accederemos así a toda la información sobre la secuencia. Lo cual siempre es interesante. Sin embargo, si queremos realizar una búsqueda rápida, lo mejor es clicar sobre la opción «FASTA» que encontramos debajo de los Accession numbers i gi (éstos también nos servirían para hacer la comparación).
Inciso: La búsqueda de secuencias proteicas (de aminoácidos) funciona de la misma manera, aunque, evidentemente, la búsqueda la tenemos que realizar en la base de datos Protein.
Volvamos a nuestro FASTA.
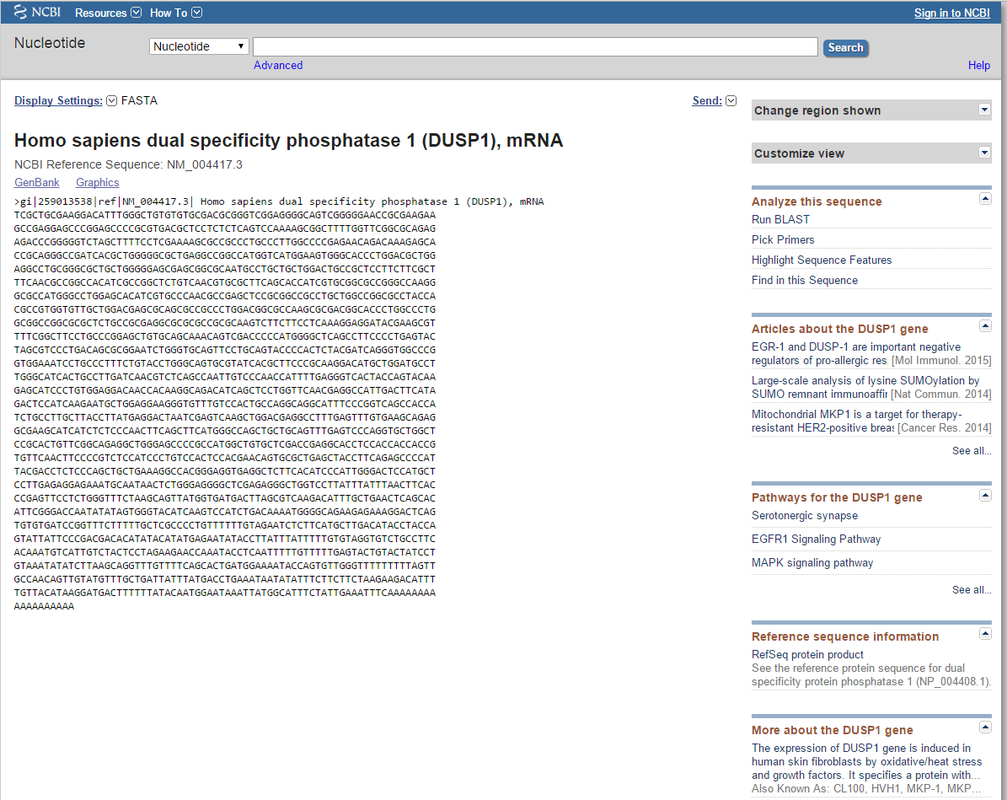
Podemos leer la secuencia de nucleótidos (A, C, G, T) del mRNA de la MKP-1 humana. A partir de aquí podríamos copiar la secuencia para realizar la comparación masiva desde la página principal del BLAST de nucleótidos.
Sin embargo, los tiempos avanzan que es un primor, y he visto que desde la propia página del FASTA podemos acceder al Blast: en el menú de la derecha: Analyze this sequence / Run BLAST. Esta acción nos lleva al mismo sitio, pero con el Accession number ya introducido en el cuadro de búsqueda.
Sin embargo, los tiempos avanzan que es un primor, y he visto que desde la propia página del FASTA podemos acceder al Blast: en el menú de la derecha: Analyze this sequence / Run BLAST. Esta acción nos lleva al mismo sitio, pero con el Accession number ya introducido en el cuadro de búsqueda.
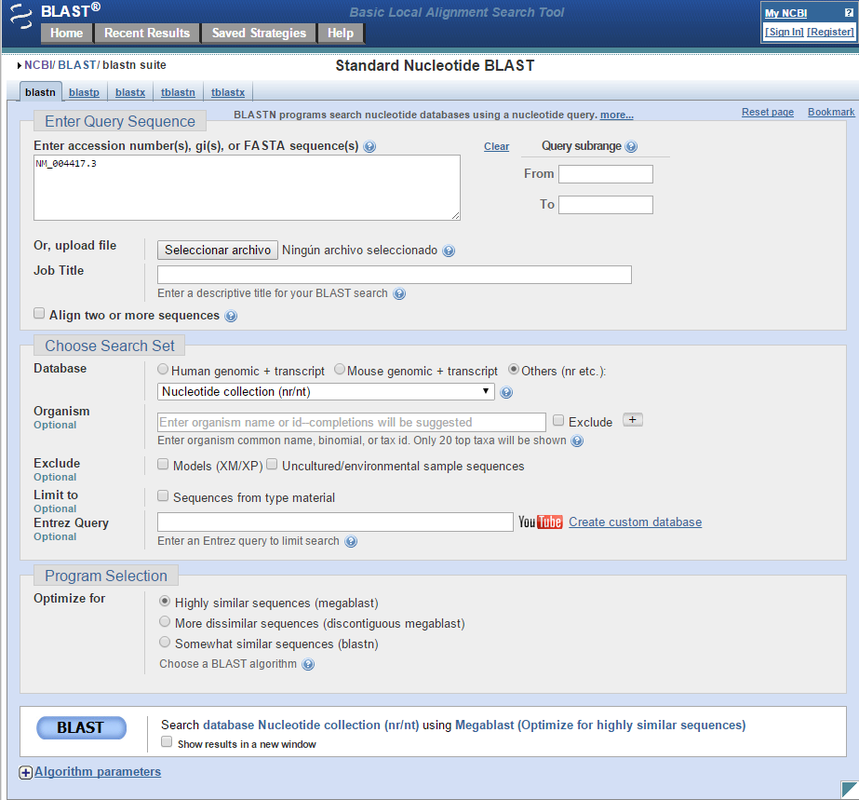
En esta pantalla podemos modificar diferentes parámetros (como, por ejemplo, los algoritmos BLAST que escogemos en el «Program Selection» desde el megablast hasta el blastn). En este ejemplo, utilizaremos el blastn.
Debajo del botón de BLAST (que daría inicio a la búsqueda), podemos observar el desplegable «Algorithm parameters». En este ejemplo cambiaré el Max Target Sequence a 1.000.
Cuando tenemos los parámetros que deseamos, clicamos el botón de BLAST (recomiendpo escoger antes la casilla Show results in a new window) y esperamos viendo páginas como la siguiente:
Debajo del botón de BLAST (que daría inicio a la búsqueda), podemos observar el desplegable «Algorithm parameters». En este ejemplo cambiaré el Max Target Sequence a 1.000.
Cuando tenemos los parámetros que deseamos, clicamos el botón de BLAST (recomiendpo escoger antes la casilla Show results in a new window) y esperamos viendo páginas como la siguiente:
Al final nos acaba apareciendo la pantalla principal de los resultados.
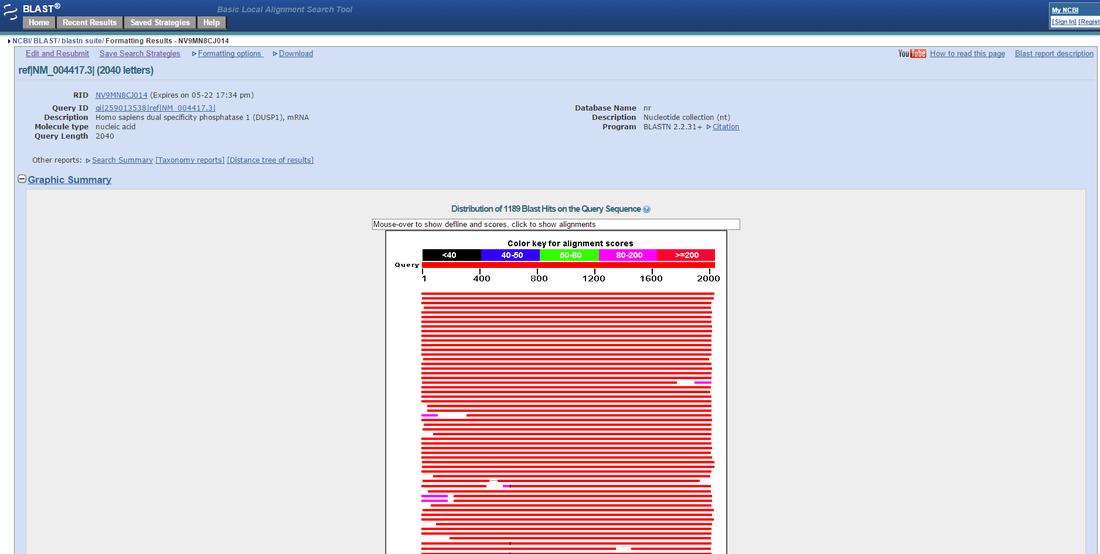
En el desplegable superior «Formating options» podemos escoger cuántos resultados se muestran en la gráfica, la extensión de cada línea, etc.
En Download nos podemos decargar los resultados (imprescindible, por cierto) en diferentes formatos (cada uno de ellos con su propia utilidad).
En el gráfico se resumen de manera visual los resultados. La leyenda se encuentra en la parte superior, sobre la escala (en este caso de unos 2.000 pares de bases). El rojo indica resultado muy, muy parecidos. Los colores hacia la izquierda indican resultados progresivamente menos certeros (con más diferencias en las secuencias).
Los resultados se ordenan por similitud. Por eso la parte superior del gráfico siempre es predominantemente roja.
Si bajamos por el gráfico, el panorama cambia:
En Download nos podemos decargar los resultados (imprescindible, por cierto) en diferentes formatos (cada uno de ellos con su propia utilidad).
En el gráfico se resumen de manera visual los resultados. La leyenda se encuentra en la parte superior, sobre la escala (en este caso de unos 2.000 pares de bases). El rojo indica resultado muy, muy parecidos. Los colores hacia la izquierda indican resultados progresivamente menos certeros (con más diferencias en las secuencias).
Los resultados se ordenan por similitud. Por eso la parte superior del gráfico siempre es predominantemente roja.
Si bajamos por el gráfico, el panorama cambia:

Estos son los últimos resultados de los 500 primeros. Podemos apreciar como los resultados presentan cada vez más zonas discontinuas y zonas de colores alejados del «confortable» rojo.
Al pasar por encima de cada resultado nos informa de qué secuencia es la que estamos observando.
Por debajo de la gráfica encontramos los resultados textuales, con algunos datos numéricos (como el % de similitud).
Al pasar por encima de cada resultado nos informa de qué secuencia es la que estamos observando.
Por debajo de la gráfica encontramos los resultados textuales, con algunos datos numéricos (como el % de similitud).
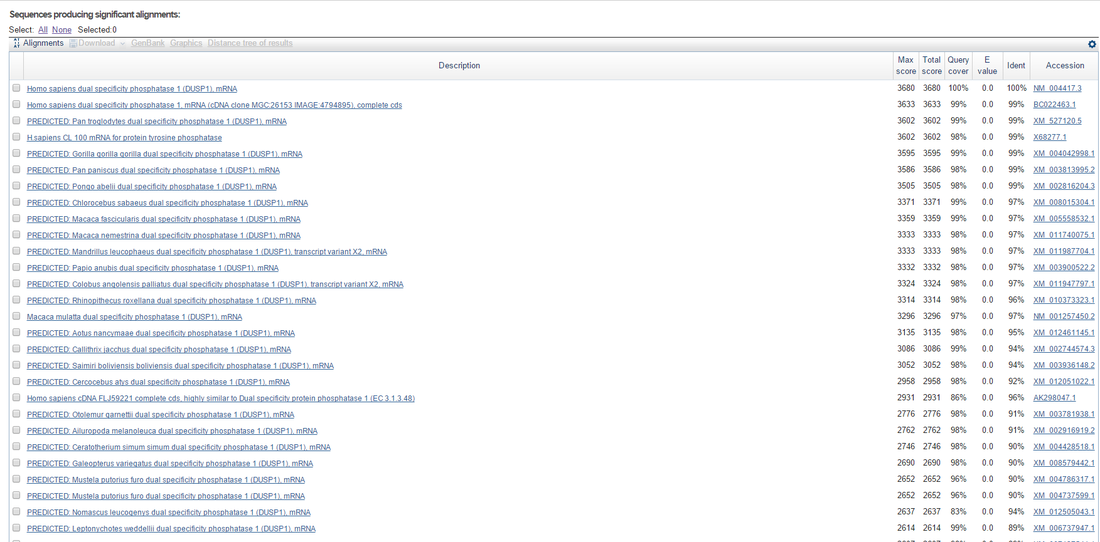
En este ejemplo ya podemos ver que nos empiezan a aparecer secuencias del mRNA de este gen de otras especies y que las que presentan mayor similitud son, lógicamente, las de humanos, chimpancés y gorilas.
Pasado esta lista de resultados nos encontramos con los resultados crudos en los que podemos ver la comparación nucleótido a nucleótido de la secuencia que hemos lanzado y cada una de las secuencias que han dado algún resultado positivo.
En la siguiente imagen, por ejemplo, podemos ver la comparación de la secuencia del mRNA de la MKP-1 humana con la del mRNA del turón europeo.
Pasado esta lista de resultados nos encontramos con los resultados crudos en los que podemos ver la comparación nucleótido a nucleótido de la secuencia que hemos lanzado y cada una de las secuencias que han dado algún resultado positivo.
En la siguiente imagen, por ejemplo, podemos ver la comparación de la secuencia del mRNA de la MKP-1 humana con la del mRNA del turón europeo.
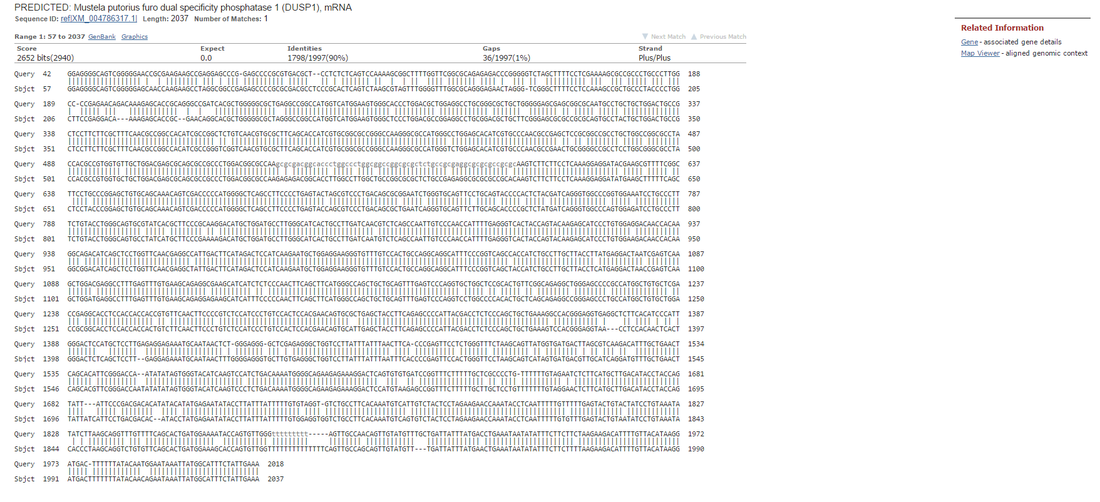
Como podéis comprobar, una cantidad de información impresionante. Pero volvamos arriba.
En la parte superior de la página de resultados nos encontramos con las opciones de «Other reports» donde podremos escoger, por ejemplo, ver el informe taxonómico o el árbol de distancias entre los resultados. Escogeremos este último; se nos abrirá una nueva pestaña en el navegador.
En la parte superior de la página de resultados nos encontramos con las opciones de «Other reports» donde podremos escoger, por ejemplo, ver el informe taxonómico o el árbol de distancias entre los resultados. Escogeremos este último; se nos abrirá una nueva pestaña en el navegador.
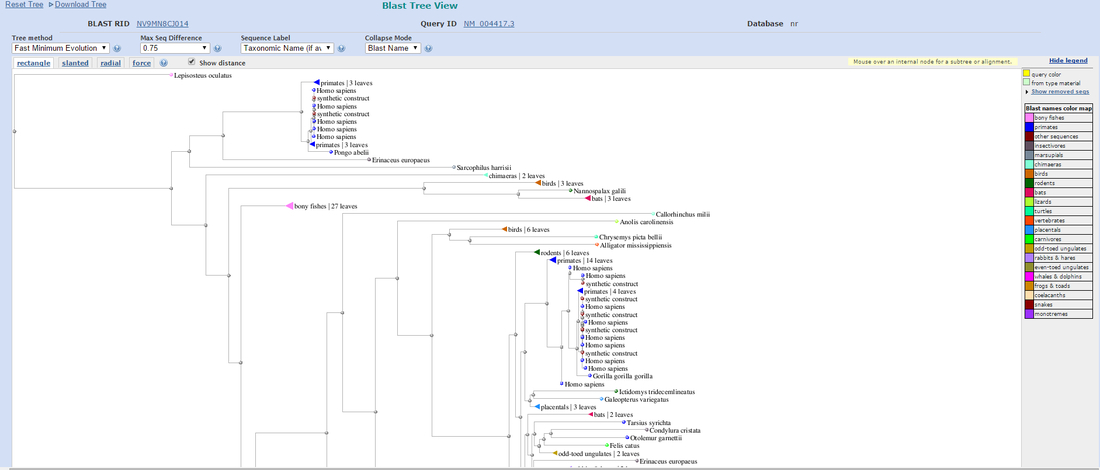
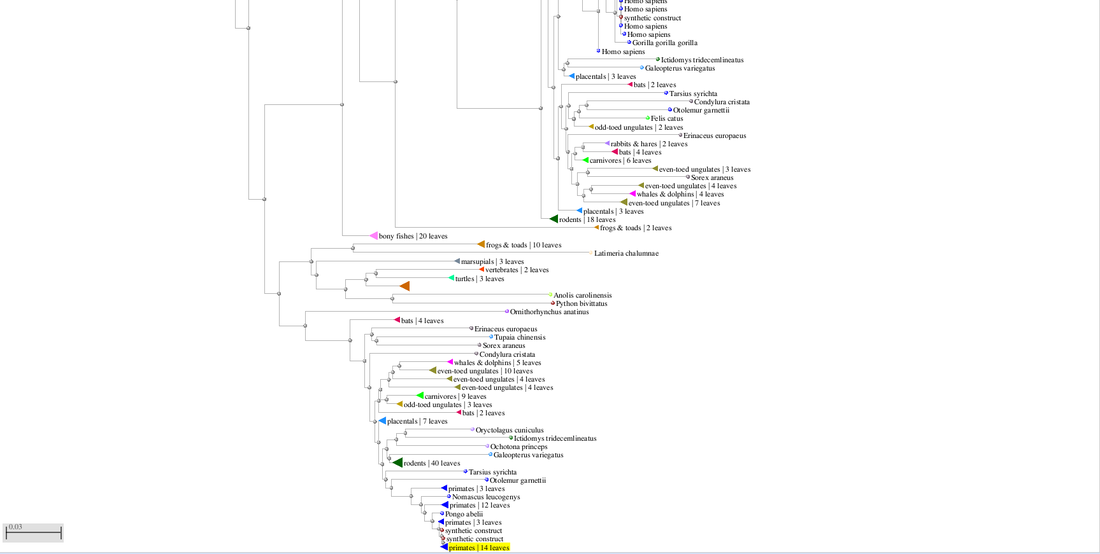
En los desplegables superiores podemos modificar parámetros de visualización (como por ejemplo si queremos o no que algunas ramas «colapsen» y se agrupen en nodos comunes).
A la derecha vemos la leyenda de colores «taxonómicos».
Y con toda esta información ya tenéis para unas cuantas horas de análisis.
Lo dicho, un trabajo que resultará de lo más interesante.
A la derecha vemos la leyenda de colores «taxonómicos».
Y con toda esta información ya tenéis para unas cuantas horas de análisis.
Lo dicho, un trabajo que resultará de lo más interesante.

 RSS Feed
RSS Feed

